1.目的
アニリンをアセチル化して、以前から鎮痛剤や染料の原料をして用いられているアセトアニリドを合成する。そして合成法、精製法や生成物分析の基本を身に付け理解する。
2.実験器具・試薬
(i)器具
300ml三角フラスコ、100ml三角フラスコ、100mlメスシリンダー、
ガラス棒、吸引ビン、アスピレーター、ヌッチェロート(ブフナーロート)、
融点測定装置、アルミボール、ガスバーナー、三脚、セラミック付き金網、
素焼板、洗浄ビン、薬さじ、スパチュラ、ミクロスパーテル、ガラス管
キャピラリーチューブ、電子天秤、5ml駒込ピペット、薬包紙
(ii)試薬
アニリン 2.5ml、無視酢酸 3.1ml、o-アセトトルイジド、
酢酸ナトリウム・三水和物 4.0g、濃塩酸 2.3ml
3.実験操作
A アセトアニリドの合成
300ml三角フラスコにメスシリンダーで採った水道水70mlを入れ、駒込ピペットを用いて濃塩酸2.3mlをフラスコに振ってかき混ぜながら入れる。これにアニリン2.5mlを駒込ピペットを用いて加え、油分がなくなるまでフラスコを振って溶かす。別に100ml 三角フラスコに酢酸ナトリウム・三水和物4.0gを入れ、水15mlで完全に溶かす。次に最初に生成させたアニリン塩酸塩に、無水酢酸3.1mlを駒込ピペットを用いて加え、よく撹拌し、予め調製しておいた酢酸ナトリウム水溶液を振りかき混ぜながらゆっくり加えよく撹拌する。最後に、水を入れたアルミボウルに300ml三角フラスコを入れて冷やし、析出した結晶を吸引ろ過する。得られたアセトアニリドは、用意した紙の上に移し、水分を取り除いた後、生成量(収量1*1)を測定する。
*1;収量1は、粗製のアセトアニリドの収量である。またここでの収率を収率1とする。
B アセトアニリドの精製
実験操作Aで得られた粗製のアセトアニリドのうち2.0gを100mlの三角フラス コに入れ、水60mlをメスシリンダーで採って加えた後に、ガスバーナーで加熱しながら完全に溶かす。油状の沈殿物があれば完全に溶解させてから加熱を止め放冷する。手で触れられるほどの温度になったら、水で冷やして再結晶させる。析出したアセトアニリドは実験操作Aと同様に吸引ろ過し、水分を取り除いた後に精製量(収量2*2)を測定する。
*2;収量2は粗製アセトアニリドから精製した量。またここでの収率を収率2とし、粗製アセトアニリド2.0gから得られた精製量の割合を示すものとする。
C 融点測定
まず、粗製の融点測定をする。ついで精製後のアセトアニリドの融点を測定する。これらの試料は、別途に用意された感想済みのものを使用する。また、混合試験も必要である。そこで反応副生成物であるo-アセトトルイド試薬と精製後のアセトアニリドを極少量ずつ等量取り、素焼き板の上でマイクロスパチュラのへら部を使い、よく混合しキャピラリーにつめ、その融点を測定する。
5.結果
(1)粗製および精製後のアセトアニリドの収量と収率
(i) アニリンがアセトアニリドになるまでの反応式
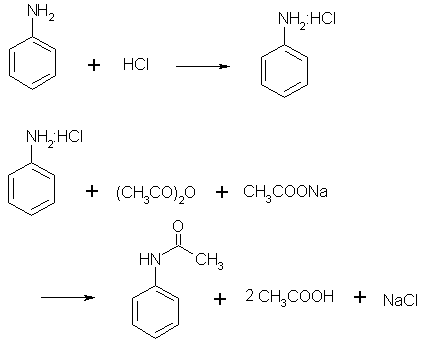
(ii) 収量と収率
(i) から実験試薬が全て等モル比であることがわかる。ここで実験に使った試薬量を物質量に換算すると、以下のようになる。ただし、アニリンとアセトアニリドノの分子量をそれぞれ93・135とする。
アニリン 2.5[ml]×1.021[g/ml]/93[g/mol]=0.0274[mol]
塩酸 2.3[ml]×0.463[g/ml]/37.5[g/mol]=0.0284[mol]
無水酢酸 3.1[ml]×1.069(g/ml)/102[g/mol]=0.0325[mol]
酢酸ナトリウム・三水和物 4.0(g)/(82+54)[g/mol]=0.0294[mol]
よって限定反応物質はアニリンになる。すなわち、アニリンと等モルのアセトアニリドできることになる。
アセトアニリドの理論収量は、3.699gである。
収量1(粗製アセトアニリド) 3.8g
収量2(精製アセトアニリド) 1.8g
収率1 3.8[g]/3.699[g]×100=102.7[%]
注)水温が10℃であった仮定すると、アニリンの溶解度は0.441である。この条件での補正を行った。
収率2 1.4[g]/(2.0[g]-0.441[g/100ml]×0.6[100ml])[g]×100
=80.67
=80.7[%]
表1. 収量1・2と収率1・2
|
|
アセトアニリドの収量 [g] |
収率 [%] |
|
粗製アセトアニリド |
3.8 |
102.7 |
|
精製アセトアニリド |
1.4 |
80.7 |
よって、実験全体の収率は下記のような計算で求めることが出来る。
{3.8[g]×1.4[g]/(2.0[g]-0.441[g/100ml])}/3.699[g]×100=82.88
=82.9[%]
すなわちこの計算は、(収率1)×(収率2)÷100である。
(iii)参考資料
|
温度(℃) |
溶解度(g/100g 水) |
|
0 |
0.360 |
|
10 |
0.441 |
|
20 |
0.561 |
|
30 |
0.729 |
|
40 |
0.975 |
|
50 |
1.33 |
|
60 |
1.86 |
|
80 |
4.5 |
|
100 |
7 |
|
120 |
13 |
|
140 |
28 |

グラフの黒い線は近似曲線である
(2)融点測定
表2. アセトアニリドの融点ならびに混合試験の結果
|
|
融け始めの温度〜完全に溶けた温度[℃] |
*3上昇にかけた時間[秒] |
|
アセトアニリド(粗製) |
113〜114 |
1620 |
|
アセトアニリド(精製) |
114〜115 |
1650 |
|
*4混合試験 |
77〜79 |
570 |
注)温度の上昇の仕方は、はじめ60℃まで素早く温度を上昇させた後、一分間で2℃の上昇速度(2℃/min.)で行った。
*3;上昇にかけた時間は、試料が完全に溶けた温度から60℃を引いた温度を上昇速度で割ったものを秒に換算したものとする。
*4;混合試験の試料は、アセトアニリドとo-アセトトルイドの1対1の混合物とする。
6.考察
(1)アセトアニリドの合成
(i)合成と収率1
粗製アセトアニリドの合成での収率が100%を超えてしまった理由として考えられる原因は大きく分けて2つある。まず1つ目は、吸引ろ過で水を十分に吸い出さなかったために、紙で水分を吸収するときに完全に吸収できなかった。本来なら、暗所に1週間ほど静置して完全に水分などを蒸発させるべきである。2つ目は、無水酢酸が過剰物質なのでアセチル基が過剰または別所に付加や置換したため、アセトアニリドより分子量が大きき化合物が出来た。主にo-アセトトルイドである。有機反応では良く起こることなので、あまり気にならなかった。ここではあまり実験操作がなかったので、実験操作の不備ではないと思う。
(ii)精製と収率2
精製で収率2が下がった原因とした考えられる3つの事柄である。まず、1つ目は実験操作に不備があったためである。アセトアニリドを煮沸して完全に溶かすときに加熱をしすぎて、アセトアニリドが少量揮発した事。2つ目に、煮沸に使った三角フラスコからブフナー漏斗に移す時に一気に入ってしまい、ろ紙上以外の場所にも多く付いてしまった。三角フラスコの内壁に結晶が多き、すべてを取る事ができなかった。3つ目に、(i)で挙げたように水溶性の反応副生成物が多く出来ていた。実験全体での収量は83%となり、かなり高い収量であった。文献によれば、同方法で収率が80%くらいまで高められる。実験操作がとてもうまくいったのかもしれない。出なければ、性質が似た副生成物が多かったのだろう。
(2)融点測定
実験結果で示したように、混合試験以外は融点温度が1℃以内である。誤差1℃以内であることから、この測定はほぼ正確に行われたと考えられる。また、粗製アセトアニリドが精製アセトアニリドより融点が低いのは、言うまでのないが不純物が含まれているからである。しかし粗製のアセトアニリドの融点の誤差が、精製したアセトアニリドと1℃しか変わらなかったことから粗製のアセトアニリド純度はそれなりに高いことがわかる。精製したアセトアニリドが文献値の融点115℃と異なるのは、精製したがアセトアニリドが100%でないためであると考えられる。また、気圧が低かったのかもしれない。
7.課題
(1)アセトアニリドの工業的製法
(i)硫酸触媒による塩化アセチル(アセチル化剤)とアニリンの反応
(ii)アニリンを2当量の酢酸と加熱する反
(iii)アニリンと酢酸に少量のベンゼンを添加し、生成水を共沸で取りの除きながらの反応
(iv)フッ化ホウ素触媒によるアニリンとアセトアミドの反応
(2)アセトアニリドの用法
(i)鎮痛解熱作用があるため、薬として使われる。現在はあまり用いられない。
(ii)医薬品の中間体合成に用いられる。
(iii)染料合成の原料として用いられる。
(3)アセトアニリドの検出方法
(i)エックス線や中性子線などを用いた回折分析
(ii)液体クロマトグラフィーを用いた方法
(iii)核磁気共鳴分析装置を用いた方法
(iv)塩基性物質でケン化後、アニリンの定性(さらし粉とニクロム酸カリウム)
